Cell Type-Resolved Top-Down Proteomics with Limited Biomass
Overview
One of the primary challenges in the field of proteomics is required sample amounts. The majority of proteomics workflows require large starting sample amounts. In this TR&D, we seek to create streamlined approaches and novel technologies that can allow for increased depth of coverage by profiling only biologically relevant cell types and tissues and better quantify proteoforms through the methods created in TR&D6. We outline a plan of attack involving common varieties of clinical samples: peripheral blood mononuclear cells (PBMCs) (DBP 5), brain sections (DBP 10, 12, and 13), diseased tissue (DBP 12 and 14) and solid tumors (DBP 9). Our first approach involves fluorescence-activated cell sorting (FACS) and laser capture microdissection (LCM). Both approaches require unique considerations to avoid aberrant proteoform processing during sample collection, but these new protocols will be readily transferrable to groups outside of the DBPs during the dissemination phase. The second approach is a novel, direct method for ablating intact proteins, which employs a picosecond infrared laser (PIRL). This approach allows for direct sampling from the tissue, thereby enabling improved spatial resolution for top-down proteomics. The final approach is specifically designed for combatting issues related to low sample amounts. It utilizes the newly-developed harmonic charge detection mass spectrometry (hCDMS), which monitors individual ions rather than hundreds of thousands during each data acquisition event and allows for high-resolution acquisition of small sample amounts.
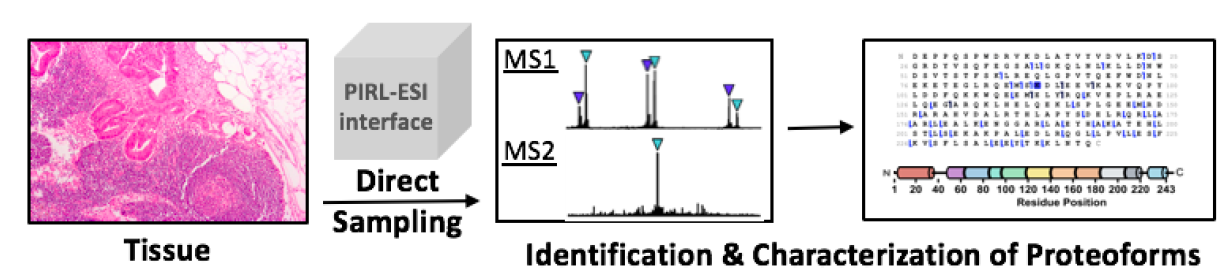
Picosecond Infrared Laser-Desorption by Impulsive Vibrational Excitation (PIRL-DIVE)
To assess the extent to which proteoform differences are present, we will focus on PIRL sampling of human primary macrophages (relevant to both DBP 5 and DBP 14), a cell type in which we have observed about 70% of the ~4,000 proteoforms identified are proteolytically degraded during sample preparation (as determined by a “biomarker” search in ProSight, which finds subsequences of full length proteoforms due to proteolysis). The open question is, “what amount of this proteolysis is endogenous and what is artefactually induced through cell lysis and our standard sample workup?” For normal B or T cells processed in the same manner, only ~20% of the hits are from biomarker search mode. To the extent PIRL-based sampling can prevent artefactual proteolysis or degradation, this experimental outcome alone will make PIRL of high interest for diverse applications in the NRTDP. For higher fidelity sampling of proteoforms and their complexes directly from soft or frozen tissue, we will therefore become better experts in this significant source of pre-analytical bias that create large differences in proteolytic state and PTMs for whole proteoforms.
Comments are closed.